

Spruce TK
The quality of results from any biophysical modeling project depends strongly on the ability to first prepare a system from an experimental data file, such as PDB or mmCIF. Unfortunately, these experiments often cannot resolve key pieces of the biological system. The missing details could be as innocent as the location of the protons, or as extreme as the missing entire mobile portions of a protein.
Spruce TK streamlines the preparation process by automatically breaking the system into individual biological components, adding any missing protons or residues, and it finishes by optimizing the hydrogen bond network for the entire system.
Spruce TK’s structure preparation workflow performs tasks including the enumeration of biological units, alternate locations (if present), modeling missing residues and loops, and placing and optimizing hydrogens, accounting for the likely tautomer states of bound heterogens (ligands and cofactors).
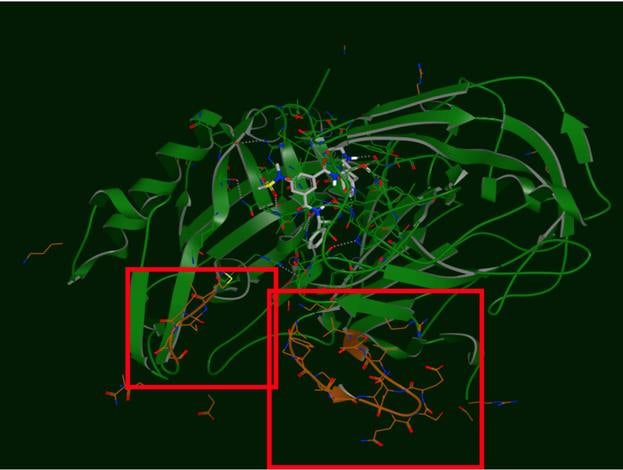
The produced output of Spruce TK is an OEDesignUnit. The OEDesignUnit has everything well componentized, making it easy to select which key pieces to include in the subsequent modeling tasks, and which to discard (e.g. excipients).
Spruce TK furthermore leverages the Iridium categorization [1] by providing the user with information about which structure is best to use for modeling. Additionally, Spruce TK highlights the parts of a structure that need special attention, if it is to be used in modeling.
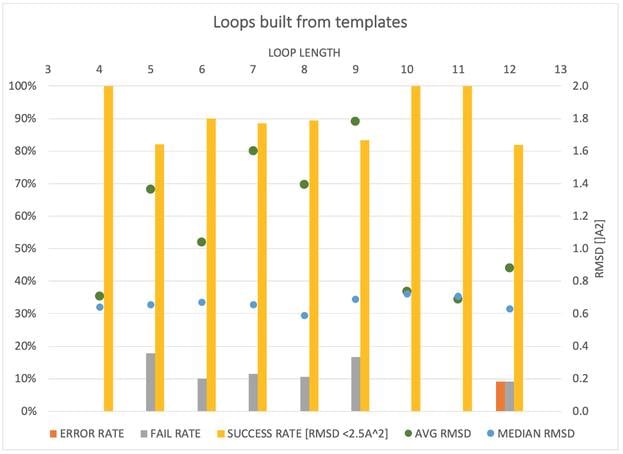
Spruce TK provides an expanding array of modeling tasks, like point-mutations, side-chain re-modeling and loop modeling using a template-based approach.
Spruce TK, furthermore, provides access to several superposition methods, based on sequence, secondary structures, or active site shape, each providing benefits depending on the similarity of the proteins being superposed.
Structure-based drug design requires careful preparation of experimental structures for downstream modeling applications. Spruce TK is a comprehensive, biomodeling preparation tool that reads experimentally solved (or modeled) protein and/or nucleic acid structures in several file formats and makes them modeling ready, for docking or molecule simulations.
To find out how Spruce TK can help with your protein modeling projects, contact us at info@eyesopen.com
Documentation
